Article Text

Abstract
Objective To investigate the role of neuroinflammation in asymptomatic and symptomatic amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) mutation carriers.
Methods The neuroinflammatory markers chitotriosidase 1 (CHIT1), YKL-40 and glial fibrillary acidic protein (GFAP) were measured in cerebrospinal fluid (CSF) and blood samples from asymptomatic and symptomatic ALS/FTD mutation carriers, sporadic cases and controls by ELISA.
Results CSF levels of CHIT1, YKL-40 and GFAP were unaffected in asymptomatic mutation carriers (n=16). CHIT1 and YKL-40 were increased in gALS (p<0.001, n=65) whereas GFAP was not affected. Patients with ALS carrying a CHIT1 polymorphism had lower CHIT1 concentrations in CSF (−80%) whereas this polymorphism had no influence on disease severity. In gFTD (n=23), increased YKL-40 and GFAP were observed (p<0.05), whereas CHIT1 was nearly not affected. The same profile as in gALS and gFTD was observed in sALS (n=64/70) and sFTD (n=20/26). CSF and blood concentrations correlated moderately (CHIT1, r=0.51) to weak (YKL-40, r=0.30, GFAP, r=0.39). Blood concentrations of these three markers were not significantly altered in any of the groups except CHIT1 in gALS of the Ulm cohort (p<0.05).
Conclusion Our data indicate that neuroinflammation is linked to the symptomatic phase of ALS/FTD and shows a similar pattern in sporadic and genetic cases. ALS and FTD are characterised by a different neuroinflammatory profile, which might be one driver of the diverse presentations of the ALS/FTD syndrome.
- neuroinflammation
- amyotrophic lateral sclerosis
- frontotemporal dementia
- GFAP
- cerebrospinal fluid
- CHIT1
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are rare neurodegenerative diseases and thought to represent different manifestations of a single disease syndrome with shared pathogenesis.1–3 To date, the cause of ALS and FTD is unclear for most (ie, sporadic) cases (sALS and sFTD) except for 5%–10% of patients with a clear monogenic background (gALS, gFTD) and there is no disease-modifying treatment option available.4 5 The most prevalent identified gene mutation in both disease entities is a large intronic GGGGCC-hexanucleotide expansion in C9orf72. Other genes affected in gALS or gFTD include SOD1, FUS, TARDBP, TBK1, NEK1, MAPT and GRN.4 6
Neuroinflammation is an important hallmark of ALS and FTD (reviewed in refs 7 8). Activated microglia and astrocytes are observed in the central nervous system (CNS) of patients with ALS/FTD by immunohistochemistry9 10 and PET imaging.11 Also preclinical disease models show substantial microglia activation and astrogliosis and the inhibition of inflammatory pathways is beneficial.7 However, anti-inflammatory drugs failed so far in human clinical trials12 and the contribution of neuroinflammation to the diseases is still controversial. Especially, its temporal role is unclear and relies largely on transgenic animal models8 and a direct comparison of the neuroinflammatory pattern between ALS and FTD is also missing. To study the preclinical phase of ALS/FTD in human individuals, we recently established a cohort of asymptomatic ALS and FTD gene mutation carriers and we could successfully show that axonal degeneration increases massively with disease onset.13
In the present study, we used the inflammatory marker proteins chitotriosidase 1 (CHIT1), chitinase-3-like protein 1 (CHI3L1, YKL-40) and glial fibrillary acidic protein (GFAP) in cerebrospinal fluid (CSF) to characterise neuroinflammation in asymptomatic and symptomatic ALS/FTD gene mutation carriers to get a hint about the temporal initiation of neuroinflammation in ALS/FTD. We selected these three markers because recent studies by our group14 and other groups15–17 consistently showed increased concentrations of CHIT1 and YKL-40 in sALS supporting their reliability. GFAP is a brain-specific protein and an established marker of astrogliosis. In addition to the temporal role, we compared the neuroinflammatory profile between patients with ALS and FTD and also between sporadic and genetic cases.
Materials and methods
Patients
The genetic patient cohort (table 1) consisted of patients with gALS recruited from the Department of Neurology of the Ulm University Hospital and University of Umeå and patients with gFTD (all with the behavioural variant of FTD, bvFTD) enrolled at different clinical centres of the German FTLD consortium (Ulm, Munich, Erlangen, Homburg, Bonn, FTLDc-TRACE study). Asymptomatic first-degree relatives of patients with familial ALS were recruited via the German Presymptomatic-ALS cohort.13 First-degree relatives without a mutation were assigned to the control group, and mutation carriers without signs of upper or lower motor neuron affection formed the group of asymptomatic ALS/FTD mutation carriers.
Characteristics of genetic ALS/FTD cohort
Patients of the sporadic patient cohort (online supplementary table S1), recruited at the Department of Neurology, Ulm University Hospital, included sALS, sFTD (all bvFTD) and control patients without neurodegenerative disease.
Supplemental material
Patients with ALS and FTD were diagnosed according to accepted criteria.18 19 All patients or their relatives gave written informed consent.
All patients underwent neuropsychological testing using standard procedures. Disease severity in patients with ALS was assessed using the ALS Functional Rating Scale-Revised (ALSFRS-R) and in patients with FTD using the FTLD-specific Clinical Dementia Rating score. Genetic testing for a panel of known ALS/FTD genes was performed according to standard protocols (details available on request). The 24 bp duplication of CHIT1 (c.1049_1072dup, NM_003465.2) was detected by size determination in agarose gel (4%) electrophoresis after PCR amplification.
CSF was collected at diagnostic evaluation by lumbar puncture (LP), centrifuged and stored within 2 hours at −80°C. Plasma (Umea cohort only) and serum samples (herein after referred to as blood samples) were treated likewise.
Postmortem spinal cord samples were obtained from five patients with gALS (two female, three male, age: 52.8±12.6 years) and one patient with gFTD (male, age: 75 years) carrying C9orf72 mutations and five neurological controls (two female, three male, age: 57.6±10.1 years) with the following diagnosis: (1) polyradiculitis and toxic myopathy; (2) subdural haematoma, haemorrhagic infarct and multiple sclerosis; (3) cerebral microangiopathy; (4) subdural haematoma, intracerebral bleeding and small vessel disease; and (5) argyrophilic grain disease and subcortical vascular encephalopathy.
Biomarker determination in CSF, blood and spinal cord tissue
CSF and blood concentrations of CHIT1 were measured using an ELISA from MBL (Belgium).15 YKL-40 was measured with the MicroVue ELISA from Quidel (USA).20 GFAP in CSF was determined with an ELISA from BioVendor (Czech Republic) and GFAP in blood was measured with the Simoa GFAP Discovery Kit (Quanterix, USA). Neurofilaments (neurofilament light chain, NfL; phosphorylated neurofilament heavy chain, pNfH) were measured using ELISAs from Uman Diagnostics, Sweden (NfL) and BioVendor, Czech Republic (pNfH), respectively.
CHIT1 expression in postmortem spinal cord tissue of five patients with gALS and one patient with gFTD (all with large hexanucleotide expansions in C9orf72), four non-neurodegenerative controls and one patient with multiple sclerosis was analysed with immunoblot using a rabbit-anti-CHIT1 antibody (Sigma number HPA010575). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression was used for normalisation.
Statistical analysis
Statistical analysis was performed using GraphPad Prism V.5.0. Groups were compared by Mann-Whitney test (two tailed) or Kruskal-Wallis test and Dunn’s post hoc test. Correlation analyses were performed using Spearman’s rank correlation coefficient. CSF YKL-40 and blood GFAP were age adjusted using a linear regression model. Frequencies of the CHIT1 24 bp duplication in exon 10, their deviation from Hardy-Weinberg equilibrium and of sex in the cohorts were compared by Χ2 test. Densitometric analysis of immunoblots was performed using ImageJ V.1.48 software, and CHIT1 expression in spinal cord tissue was normalised to GAPDH and compared by Student’s t-test (two tailed). A p value <0.05 was regarded as statistically significant.
Results
Chitotriosidase 1
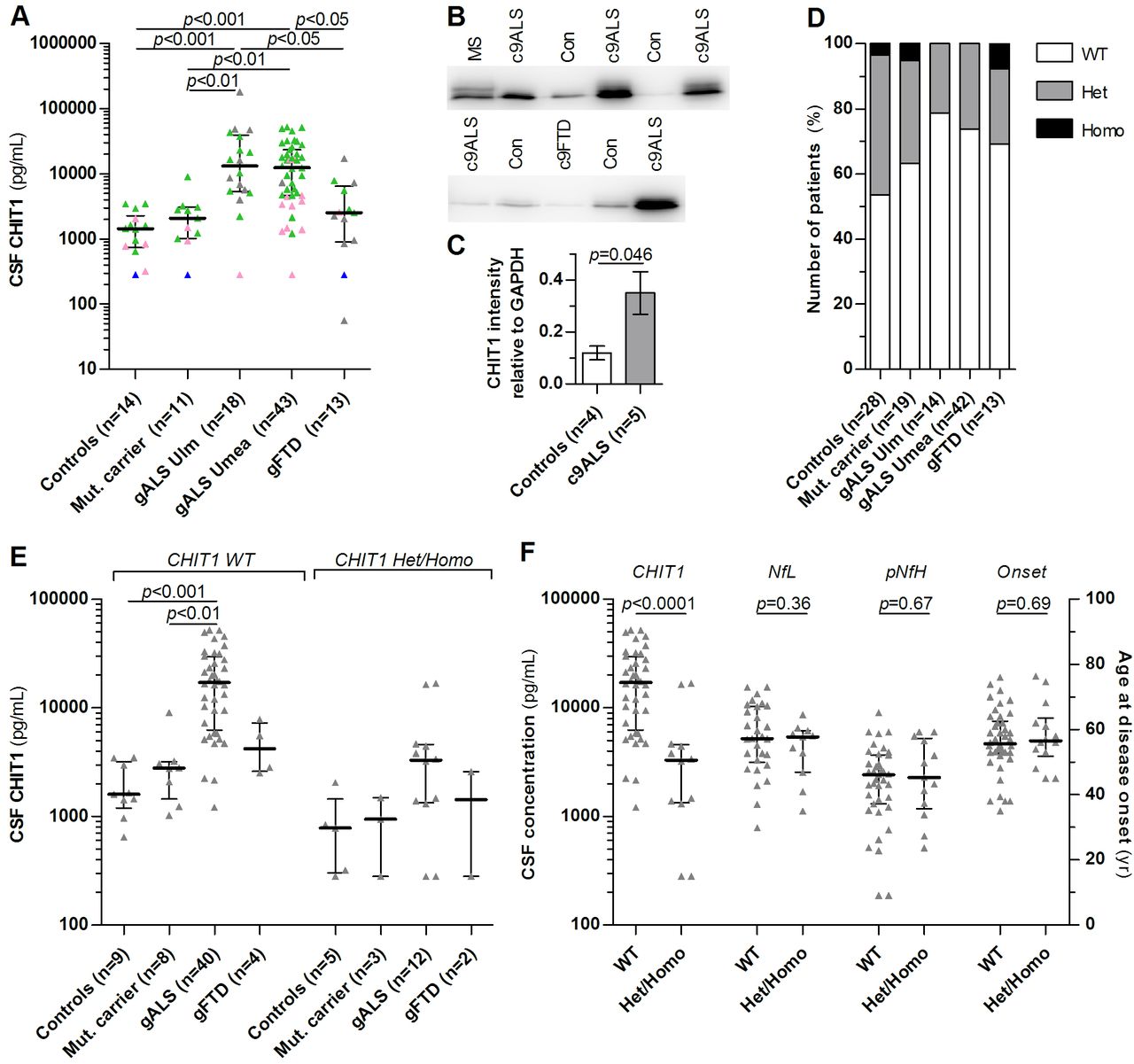
Characteristics of patients are listed in table 1 (gALS/gFTD cohort) and table S1 (sALS/sFTD cohort). The patients with gALS and gFTD were older than controls (and asymptomatic mutation carriers) (p<0.001). The median CHIT1 concentration in CSF was fourfold to ninefold increased in the patients with gALS compared with controls, asymptomatic mutation carriers and gFTD cases. The other groups did not differ statistically from one another (figure 1A). Correlation of CHIT1 concentration in CSF with age in controls was weak (r=0.37) and not significant (p=0.11). No significant differences of CHIT1 were observed in blood except between gALS from Ulm and controls (p<0.05, table 1). Blood and CSF concentrations of CHIT1 were significantly correlated (r=0.51, p<0.0001) and CHIT1 concentrations in CSF were strongly positively correlated with concentrations of the axonal degeneration markers NfL and pNfH (r=0.65 and r=0.69, p<0.0001). We confirmed increased CHIT1 expression also in spinal cord tissue of gALS cases by immunoblot (p<0.05) whereas no alteration was observed in the single autopsied FTD case (figure 1B,C).

CHIT1 is increased in patients with gALS but not asymptomatic mutation carriers and gFTD. (A) CHIT1 was measured in CSF by ELISA in healthy patients without ALS mutation (controls), in asymptomatic ALS/FTD mutation carriers (Mut carrier), in two independent cohorts (Ulm and Umeå) of patients with genetic ALS (gALS), and in patients with genetic FTD (gFTD) suffering from the behavioural variant of FTD (bvFTD). Bars and whiskers are median and IQR, triangles are individual values. Colours indicate the status of the 24 bp duplication of CHIT1: no duplication (green), heterozygous (pink), homozygous (blue), no information (grey). (B) CHIT1 immunoblots from postmortem spinal cord tissue of patients with ALS and FTD carrying the C9orf72 mutation (c9ALS and c9FTD), multiple sclerosis (MS) and non-neurodegenerative controls and (C) quantitative comparison of CHIT1 expression relative to GAPDH (mean±SD) using Student’s t-test. (D) Frequency of the 24 bp dublication of CHIT1 in the disease groups, p=0.38 (Χ2 test). (E) CHIT1 concentration in CSF in the disease groups depending on CHIT1 genotype. (F) Comparison of CHIT1, NfL and pNfH concentrations in CSF and age at disease onset in patients with gALS (Ulm+Umeå) with or without the 24 bp duplication of CHIT1. Heterozygous (Het), homozygous (Homo), no duplication (WT). Bars and whiskers are median and IQR, triangles are individual values. Groups were compared by Kruskal-Wallis test and Dunn’s post hoc test. ALS, amyotrophic lateral sclerosis; CSF, cerebrospinal fluid; FTD, frontotemporal dementia; NfL, neurofilament light chain; pNfH, phosphorylated neurofilament heavy chain.
A 24 bp duplication in exon 10 of the CHIT1 gene has been described previously resulting in reduced expression and activity of CHIT1.21 Because the prevalence of this polymorphism is high in European populations (35%–50%),22 we genotyped our genetic patient cohort to rule out differences in CHIT1 concentrations due to CHIT1 genotypes. About 43% (n=12) and 4% (n=1) of control patients were either heterozygous or homozygous carriers of the 24 bp duplication of CHIT1 (figure 1D), and the affected individuals are also highlighted in figure 1A. In all groups, no significant difference of the CHIT1 genotype frequency to the Hardy-Weinberg equilibrium was observed. There was a tendency towards a lower frequency of this CHIT1 mutation in gALS cases from both centres, but this difference was not significant (p=0.38). CHIT1 concentrations were lower in CSF and serum in heterozygous and homozygous carriers of the 24 bp duplication. Notably, CHIT1 concentrations were increased in CSF in patients with gALS independently from the CHIT1 genotype (figure 1E).
Patients with gALS carrying the CHIT1 24 bp duplication had significantly lower CHIT1 concentrations in CSF compared with non-carriers, but neurofilament concentrations and age at disease onset were not affected by the CHIT1 polymorphism (figure 1F).
YKL-40
In agreement with previous studies,23 24 YKL-40 showed a moderate correlation with age in CSF of control patients (r=0.41, p=0.01) but not in serum (r=0.17, p=0.38), therefore only CSF values of YKL-40 were age adjusted. CSF YKL-40 was significantly increased in gALS and gFTD compared with controls and asymptomatic mutation carriers (figure 2A) by a factor of 2–5. There was no difference between controls and asymptomatic mutation carriers and between gALS and gFTD. We did not observe alterations in blood YKL-40 (p=0.36, table 1) and there was only a weak correlation between CSF and blood concentrations (r=0.30, p=0.0053). Since data about YKL-40 in CSF of patients with sALS are limited, we investigated an additional cohort of age (p=0.88) and sex-matched (p=0.91) patients with sALS and sFTD. Both patients with sALS and sFTD had increased concentration of YKL-40 in CSF with slightly higher values in sFTD compared with sALS (p<0.05, figure 2B). Correlation analysis showed a strong correlation of CSF YKL-40 concentrations with the axonal degeneration markers NfL (r=0.73, p<0.0001) and pNfH (r=0.71, p<0.0001), a weak correlation with the ALSFRS-R score (r=−0.42, p<0.0001) and no correlation with disease duration at LP (r=−0.18, p=0.11).

Increased levels of YKL-40 in ALS/FTD and GFAP in FTD but not asymptomatic mutation carriers. YKL-40 and GFAP were measured by ELISA in CSF of (A, C) healthy patients without ALS mutation (controls), in asymptomatic ALS/FTD mutation carriers (Mut carrier), in two independent cohorts (Ulm and Umeå) of patients with genetic ALS (gALS), and in patients with genetic FTD (gFTD) suffering from the behavioural variant of FTD (bvFTD) and (B, D) in CSF of control patients without neurodegenerative disease (controls), patients with sporadic ALS and sporadic bvFTD (sALS and sFTD). Bars and whiskers are median and IQR, triangles are individual values. Groups were compared by Kruskal-Wallis test and Dunn’s post hoc test. ALS, amyotrophic lateral sclerosis; CSF, cerebrospinal fluid; FTD, frontotemporal dementia; GFAP, glial fibrillary acidic protein.
Glial fibrillary acidic protein
The GFAP concentration in CSF was significantly increased in gFTD compared with controls (p<0.05) and gALS (p<0.05, figure 2C). There was no difference between controls, asymptomatic mutation carriers and gALS groups. Blood concentrations of GFAP did not differ between the groups studied (table 1). However, blood concentrations showed a strong correlation with age in control samples (r=0.70, p<0·0001) and therefore they were age adjusted. GFAP in CSF did not significantly correlate with age (r=0.28, p=0.05). We observed a weak correlation between blood and CSF GFAP (r=0.39, p=0.0002). For comparison with the genetic cases, we also analysed a larger group of age (p=0.88) and sex-matched (p=0.83) sALS and sFTD cases. Similarly, higher CSF GFAP concentrations were observed in patients with sFTD compared with sALS (p<0.001) and controls (p<0.01, figure 2D). The correlation of CSF GFAP with NfL and pNfH in the whole genetic cohort was very weak (r=0.04, p=0.73 and r=0.05, p=0.61) and also weak in the gFTD group (r=0.32, p=0.34 and r=0.28, p=0.38). There was no significant correlation of CSF GFAP with the ALSFRS-R score (r=−0.07, p=0.54) and disease duration at LP (r=0.10, p=0.40) in patients with ALS.
Correlation analysis and time course estimation
Correlation analysis of the three inflammatory markers in CSF yielded a moderately strong correlation of CHIT1 with YKL-40 (r=0.52, p<0.0001) and very weak correlation of CHIT1 and YKL-40 with GFAP (r=0.14, p=0.28 and r=0.09, p=0.46).
To simulate a presymptomatic time span, we used the parental age of onset to estimate the time to disease onset as previously described.13 The estimated time course of CHIT1, YKL-40 and NfL is shown in figure 3 for the asymptomatic mutation carriers and gALS cases. Here, a sudden increase of CHIT1 and YKL-40 with symptom onset, similar to the previously reported neurofilaments,13 is seen.

{kind=link}
{kind=link}
{kind=link}
Estimated time course of biomarkers in cerebrospinal fluid (CSF). Disease duration at the time of lumbar puncture is shown to estimate the time course of CHIT1, YKL-40 and NfL in CSF of asymptomatic mutation carriers and patients with genetic amyotrophic lateral sclerosis (ALS). The concentrations of CHIT1, YKL-40 and NfL were normalised to the respective mean concentration of the asymptomatic mutation carriers to allow a better comparison of the magnitude of changes. The assumed time to disease onset in the asymptomatic mutation carriers was estimated using the parental age of onset. Two cases were already older than their affected relatives and for this graph we assumed that they will have their disease onset within 6 months. One individual with SOD1 mutation showed early electromyography (EMG) abnormalities and was defined as time point 0 month. NfL, neurofilament light chain.
Discussion
The identification of early pathophysiological events in ALS and FTD is challenging because diagnosis is based on clinical symptoms18 19 making presymptomatic disease phases inaccessible to researchers. Studies of ALS/FTD mutation carriers offer a unique opportunity to investigate the critical preclinical phase because they can be identified and followed before symptom onset.13 25 We measured the microglia and astroglia markers CHIT1, YKL-40 and GFAP in CSF from asymptomatic mutation carriers and observed no significant differences of these markers compared with controls. The strong correlation of CHIT1 and YKL-40 in CSF with the axonal degeneration markers NfL and pNfH further supports a close link between neuroinflammation and the degenerative phase of the diseases. Additionally, in one individual with SOD1 mutation—only showing first electromyography abnormalities and therefore representing the transition from the presymptomatic to the symptomatic phase—increased neurofilament levels but normal CHIT1 (no CHIT1 polymorphism) and slightly increased YKL-40 (203 ng/mL) were measured. This observation in this single individual could be additional evidence that axonal damage precedes neuroinflammation in ALS, but clearly awaits further confirmation. Overall, these findings suggest that neuroinflammation—as evaluated by this panel of established neuroinflammatory markers—is either not an early event in ALS and FTD or a different kind of neuroinflammation not reflected in changes in CHIT1, YKL-40 or GFAP is taking place in the initial stages of these diseases. However, our clinical findings are in agreement with studies in transgenic human mutant SOD1 mouse models. Here also the initiation of microglia activation and astrogliosis occurs around symptom onset or even later.26
The observed increase of CHIT1 and YKL-40 levels in CSF of patients with gALS indicates profound neuroinflammation in the symptomatic phase of ALS. Our finding in gALS is in agreement with the previously reported CHIT1 increase in CSF of patients with sALS by our group14 and other groups.15–17 In this context, our observation that the 24 bp duplication in exon 10 of CHIT1, leading to lower CHIT levels,21 did not affect axonal degeneration and disease severity in patients with ALS indicates that CHIT1 itself is just a marker of the neuroinflammatory process and does not actively contribute to it. We also showed that YKL-40 is increased in both gALS and sALS which has been described only for sALS before.16 17 27 In contrast, GFAP levels in CSF are not altered in gALS and sALS. Overall, our results show that the neuroinflammatory profile investigated here is similar in gALS and sALS and may indicate that neuroinflammation is a shared pathophysiological process in both gALS and sALS.
The increased levels of YKL-40 and GFAP in patients with FTD also indicate profound neuroinflammation in the symptomatic phase of FTD. Our results support previous observations of increased YKL-4023 24 and GFAP28 in CSF of patients with sFTD and we could demonstrate similar changes in gFTD. Thus, our data indicate a similar neuroinflammatory profile in gFTD and sFTD and support the notion that it is a shared mechanism in gFTD and sFTD pathophysiology.
In contrast to CSF, no significant alterations of YKL-40 and GFAP were observed in blood. YKL-40 is not a CNS-specific protein and blood levels are also influenced by other tissues. Changes of YKL-40 in the CNS might have a lower impact on blood than on CSF levels. GFAP levels and group differences in blood are lower than in CSF. This is also known from other CNS-derived proteins such as NfL13 and could explain the missing significance in blood.
Although the increased CSF concentrations of the inflammatory markers studied here support profound neuroinflammation in both ALS and FTD, we observed significant differences in the neuroinflammatory profile between the diseases. Increased CSF levels of CHIT1 are characteristic of ALS whereas GFAP is increased in FTD only. YKL-40 is increased in both although higher levels were observed in sFTD but with considerable overlap. The selective increase of GFAP in FTD and not ALS also explains why GFAP did not correlate with neurofilaments, in contrast to CHIT1 and YKL-40, because a massive increase in CSF neurofilaments is characteristic for ALS. CHIT1 is thought to be a marker of microglia/macrophage activation14 and the increased CSF levels in ALS are also in agreement with the observed peripheral monocyte and macrophage activation in ALS.29
GFAP and YKL-40 in CSF are both considered to be markers of astrogliosis27 30 and thus, it is surprising that they behave differently in ALS and FTD. Interestingly, we observed only a very weak correlation of their CSF concentrations which is in agreement with a previous report.31 This could indicate that different astrocyte subpopulations or a different spatial distribution are reflected by GFAP and YKL-40. Overall, the elevated GFAP concentration in FTD indicates a higher degree or different type of astrogliosis compared with ALS. GFAP determination in CSF might be an additional tool to improve identification of FTD in patients with ALS. Nevertheless, follow-up studies are needed to evaluate the diagnostic potential of GFAP to detect FTD among patients with ALS.
Limitations of the study: The samples used here were obtained from longitudinal studies where neuropathological data were not yet available. Especially sFTD cases are characterised by different types of neuropathology (eg, tau or TDP43 pathology5) and this heterogeneity could have influenced our observations (considering that sALS is mainly associated with TDP43 pathology3).
It is also of note that 40% of the asymptomatic mutation carriers carry mutations related to ALS only (7x SOD1, 2x FUS, 1x TARDBP). Thus, ALS-related effects might be overestimated and FTD-related effects underestimated. Since we did not observe significant alterations in the asymptomatic mutation carriers only the last case could be relevant.
In conclusion, our data from asymptomatic mutation carriers indicate that neuroinflammation is linked to the symptomatic phase of ALS and FTD, which is in agreement with preclinical studies in mice. We show that neuroinflammation is a shared mechanism in sporadic and genetic forms of both diseases supporting the use of mutation-based animal models to study neuroinflammatory mechanisms. ALS and FTD are characterised by a different neuroinflammatory pattern with more severe macrophage/microglia activation in ALS and astrocytosis in FTD. These differences might be one driver for the manifestation of the ALS/FTD syndrome as FTD or ALS.
Acknowledgments
We are grateful to all study participants. We thank Mehtap Bulut, Sandra Hübsch, Stephen Meier, Alice Beer and Dagmar Schattauer for their excellent technical assistance.
References
Footnotes
Contributors Design or conceptualisation of the study: PO, PW, JHW, ACL, MO. Analysis or interpretation of data: PO, PW, PS, SAS, FN, AEV, JDS, PMA, HJ, JK, AD, KFa, KFl, ML, KM, AK, JP, AS, DRT, DYH, JHW, ACL, MO. Drafting or revising the manuscript for intellectual content: PO, PW, PS, SAS, FN, AEV, JDS, PMA, HJ, JK, AD, KFa, KFl, ML, KM, AK, JP, AS, DRT, DYH, JHW, ACL, MO.
Funding This study was supported by the EU Joint Programme–Neurodegenerative Disease Research (JPND) networks SOPHIA (01ED1202A), BiomarkAPD (01ED1203F), and PreFrontAls (01ED1512), the German Federal Ministry of Education and Research (FTLDcO1GI1007A, MND-Net 01GM1103A), the EU (NADINE 246513, FAIR-PARK II 633190,STRENTH/JPND), the German Research Foundation/DFG (VO2028, SFB1279), the foundation of the state Baden-Württemberg (D3830), Boehringer Ingelheim Ulm University BioCenter (D5009), Thierry Latran Foundation, Departmental Funds (GPS-ALS Study), the Charcot Foundation, the Swedish Brain Research Foundation, the Bertil Hållsten Foundation, the Swedish Research Council, the Knut and Alice Wallenberg Foundation, and the Ulla-Carin Lindquist Foundation.
Disclaimer The funders were not involved in the study design, in the collection, analysis and interpretation of data, in the writing of the report; and in the decision to submit the paper for publication.
Competing interests DRT received consultancies from Covance Laboratories (UK) and GE Healthcare (UK), received a speaker honorarium from GE Healthcare (UK), and collaborated with Novartis Pharma Basel (Switzerland).
PO, PW, PS, SAS, FN, AEV, JDS, PMA, JK, AD, KFa, KFl, HJ, ML, KM, AK, JP, AS, DYH, JHW, ACL and MO report no competing interests.
Patient consent Not required
Ethics approval Ethics committees of the Universities of Ulm, Umea, Munich, Erlangen, Homburg, Bonn. The Medical Ethical Review Boards of the participating centers approved the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial commentary