Article Text

Abstract
Introduction Achilles tendinopathy (AT) is a common and disabling musculoskeletal condition. First-line management involving Achilles tendon loading exercise with, or without, other modalities may not resolve the problem in up to 44% of cases. Many people receive injections. Yet there are no injection treatments with demonstrated long-term efficacy. The aim of the trial is to examine the 12-month efficacy of high-volume injection (HVI) with corticosteroid and HVI without corticosteroid versus sham injection among individuals with AT.
Methods and analysis The trial is a three-arm, parallel group, double-blind, superiority randomised controlled trial that will assess the efficacy of HVI with and without corticosteroid versus sham up to 12 months. We will block-randomise 192 participants to one of the three groups with a 1:1:1 ratio, and both participants and outcome assessors will be blinded to treatment allocation. All participants will receive an identical evidence-based education and exercise intervention. The primary outcome measure will be the Victorian Institute of Sport Assessment – Achilles (VISA-A) at 12 months post-randomisation, a validated, reliable and disease-specific measure of pain and function. Choice of secondary outcomes was informed by core outcome domains for tendinopathy. Data will be analysed using the intention-to-treat principle.
Ethics and dissemination Ethics approval was obtained via the Monash University Human Ethics Committee (no: 13138). The study is expected to be completed in 2024 and disseminated via peer review publication and conference presentations.
Trial registration number Australia and New Zealand Clinical trials registry (ACTRN12619001455156)
- Achilles tendinopathy
- injection
- high volume injection
- randomised controlled trial
Data availability statement
Data will be made available on request at the completion of the trial.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known
There are no injection treatments for Achilles tendinopathy with demonstrated long-term efficacy compared with placebo or sham.
The high-volume injection (HVI) includes local anaesthetic and saline with or without corticosteroid.
There is efficacy compared with sham at 6 months for the injection with but not without corticosteroid.
What are the new findings
The proposed trial will address a knowledge gap by evaluating the efficacy of the HVI with and without corticosteroid versus sham for Achilles tendinopathy over 12 months.
This research has the potential to provide evidence to support or refute the HVI in clinical practice for people with non-responsive Achilles tendinopathy
Introduction
Achilles tendinopathy (AT) is a common, painful and disabling musculoskeletal condition characterised by local tissue pathology, swelling and activity-related pain. A 2011 Dutch general practice study among 57 725 adults (21–60 years) reported an Achilles tendinopathy incidence of 2.35 per 1000 GP registered patients,1 with 35% of cases not involving sport.1 Up to 18% of running injuries are related to Achilles tendinopathy.2 The cumulative prevalence in the general population has been reported to be 6%.3 Achilles tendinopathy can severely impact quality of life (particularly mobility, usual activity and pain domains), is associated with reduced work capacity in 38% of people affected and accounts for substantial healthcare costs.4
The aetiology of Achilles tendinopathy is multifactorial5 and the imbalance between load demands placed on the tendon and its ability to remodel is considered a major factor.6 Other factors that are thought to increase tendon load or influence the remodelling capacity of the tendon may increase the risk of AT, including reduced plantarflexor strength, genetic profile and metabolic factors (eg, elevated cholesterol or diabetes).5 7 Once established, Achilles tendon tissue changes include matrix degradation (characterised by inferior quality and disorganised collagen), accumulation of hydrophilic proteoglycan molecules that increase bound water causing swelling, as well as ingrowth of blood vessels and nerves.6 The ingrowth of blood vessels and nerves from the deeper lying Kager’s fat pad is considered a potential pain mechanism in AT8 because biochemicals involved in pain transmission (eg, substance P) are released by in-growing nerve tissue.9 10 Thus, the ingrowth of nerves from Kager’s fat pad is a potential treatment target.
Achilles tendon loading exercise is recommended as a first-line treatment for Achilles tendinopathy in clinical practice guidelines and expert narratives.11 12 However, up to 44% of people may not respond to exercise treatments for AT.13 Up to 60% of people with AT have continued pain and disability after 5 years despite exercise interventions, and 48% will seek additional treatment including injections and surgery.14
There are currently no injection treatments with demonstrated long-term efficacy. Corticosteroid injections demonstrate short-term efficacy in improving tendinopathy pain and function, but there is well-documented symptom recurrence in the longer term.15–17 Other injections in clinical use for AT, including polidocanol, aprotinin and platelet-rich plasma injections, have been shown to be no more effective than placebo or sham,18 although a network meta-analysis found benefits compared with wait-and-see.19 An injection option that demonstrated efficacy compared with placebo or sham is likely to reduce the substantial and long-standing pain and disability, as well as costs associated with managing AT.
The high-volume injection (HVI) is a relatively new injection used to manage AT.20 This was first introduced in 2008.20 HVI typically consists of a corticosteroid and local anaesthetic in a 50 mL injection. The fluid is injected between Kager’s fat and the Achilles tendon and is hypothesised to disrupt neurovascular ingrowth from Kager’s fat to the Achilles tendon that is believed to be a key source of pain in AT.21 The proposed mechanisms of HVI include trauma or ischaemic pressure from the large volume of fluid which may impair nerve function and pain transmission20 22 and anti-inflammatory effect from the corticosteroid.
There have been several case series reporting positive effects of the HVI for AT20 23 24 but limited randomised trials until recently. Preliminary data from our team25 (2017, n=19 in each group) demonstrated benefit for primary pain and function outcome compared with sham injection at 6, 12 and 26 weeks.25 In a follow-up trial26 (2019, n=14 in each group), we compared HVI with and without corticosteroid and found that the short-term benefit (6 and 12 weeks) was absent but the medium-term benefit (26 weeks) remained when the corticosteroid was removed. A 2020 randomised trial27 (n=38 HVI without corticosteroid and n=41 sham) compared the HVI without corticosteroid and sham and found no differences in primary pain and function outcome at any timepoint up to 26 weeks (2, 6, 12 and 26 weeks). It is important to determine whether the effects of HVI with corticosteroid are maintained over 12 months or whether there is symptom recurrence as observed with other corticosteroid injections for tendinopathy.15 Further, the medium-term and long-term efficacy of HVI without corticosteroid is uncertain.
The primary aim of this trial is to examine the efficacy of HVI with corticosteroid and HVI without corticosteroid versus sham injection among individuals with AT at 12 months.
Methods and analysis
Study design
This is is a three-arm, parallel group, double-blind, superiority randomised controlled trial with a 12-month follow-up. Participants will be block-randomised into sham injection, HVI with corticosteroid or HVI without corticosteroid groups with a 1:1:1 ratio. Participants and outcome assessors will be blinded to treatment allocation. All participants will receive an identical evidence-based education and exercise intervention, which removes ethical concerns of withholding treatment. This protocol is reported in accordance with the 2013 SPIRIT guidelines.28
Setting
The trial is based in a large radiology clinic in central Melbourne, Australia (Imaging at Olympic Park (IOP)). This setting in a central location allows implementation of a previously successful strategy to recruit people with AT from throughout metropolitan Melbourne, and there is a large network of physiotherapists, sports physicians and surgeons who usually refer people with AT to IOP.
Sample size
The sample size was determined based on our primary endpoint, the Victorian Institute of Sport Assessment – Achilles (VISA-A) scale (0–100 points). Minimal clinically important difference (MCID) has not been determined for midportion AT, but prior trials have estimated MCID to be 10 points.25 29 Based on our preliminary data,25 26 we assume a 10-point benefit of HVI compared with sham at 12 months. Assuming a SD from our preliminary trial of 15.6,25 51 participants per group will provide 80% power to detect a 10-point difference between any of the three trial arms (type I error rate split to 1.67% for each of the three comparisons). We will recruit 64 participants per trial arm to allow for attrition of up to 20% at 12 months.
Eligibility criteria
Inclusion and exclusion criteria are shown in table 1.
Inclusion and exclusion criteria
Recruitment and retention
Recruitment channels include an advertising campaign in local newspapers, flyers advertising the trial placed at local community and sporting clubs, retirement villages and senior citizen centres, via a large network of clinical partners who refer people with AT to IOP, and via a paid social media campaign, including Facebook and Twitter. To improve retention, participants receive a $A100 shopping voucher on completion of 26-week and 52-week outcomes to reimburse them for costs incurred (time and travel).
Procedure
Respondents to newspaper, flyer and social media advertising, and potential participants alerted to the trial via clinical partners, will initially be screened via telephone (when they call the researchers, or receive a call back to social media, email or text contact). Potentially eligible participants will be provided with electronic study information and given at least 24 hours to determine whether they would like to proceed. Those who contact the researchers interested in participating will be booked into a baseline session at IOP. A researcher and ultrasonographer blinded to treatment allocation will assess eligibility (figure 1). Eligible participants will be asked to provide written consent and complete the baseline assessment prior to randomisation.

Participant flow through the trial.
Baseline assessment
Standard demographic characteristics (age, sex, weight, height, body mass index, ethnicity), healthcare history (medical history, surgical history, current medications) and specific AT history (site, duration, previous treatment) will be recorded at baseline. The following measures will also be assessed at baseline: (1) Achilles tendon pain map: via a computer application; (2) Pain Detect: a validated questionnaire used to detect people who have neuropathic pain30; (3) pressure pain thresholds: assessing local (Achilles tendon) and diffuse (lateral elbow) hyperalgesia; (4) expectancy/credibility: participants will be asked about how logical and successful they feel the treatment they have received will be, as well as their confidence in the treatment.31
Randomisation
Eligible participants will be randomly assigned to one of the three treatment arms with a 1:1:1 ratio using computer-generated permuted blocks of variable size (4, 8 and 12). This process will be undertaken remotely using third-party web-based randomisation (Griffith University, Gold Coast, Australia) to ensure allocation concealment. For eligible participants, a radiology nurse will use the web-based service to determine treatment allocation. Only the nurse who prepares the injection (figure 1) will be aware of treatment allocation and they will be instructed to minimise interaction with the participants.
Blinding
Procedures to ensure participant blinding include:
Preparing both the HVI and sham injections for all participants. The HVI consists of five 10 mL syringes, the first with exactly 10 mL of 0.5% bupivacaine hydrochloride and 25 mg of hydrocortisone, followed by four syringes with 10 mL each of normal saline. All five syringes will be prepared regardless of group allocation, ensuring preparation time is identical.
Using identical injection procedures for all groups. All five syringes will also be injected so treatment time is identical. For the sham group, the excess injectant will be diverted away from the patient and to a 50 mL syringe via a three-way stop cock (figure 2)
A sleeve placed over the first syringe, so the radiologist is not able to detect whether the syringe contains corticosteroid (the corticosteroid changes the colour of the injectant). Piloting has indicated that the colour of the injectant is not clearly visible from the thin injecting tube. The radiologist will be aware when sham is the allocated group.
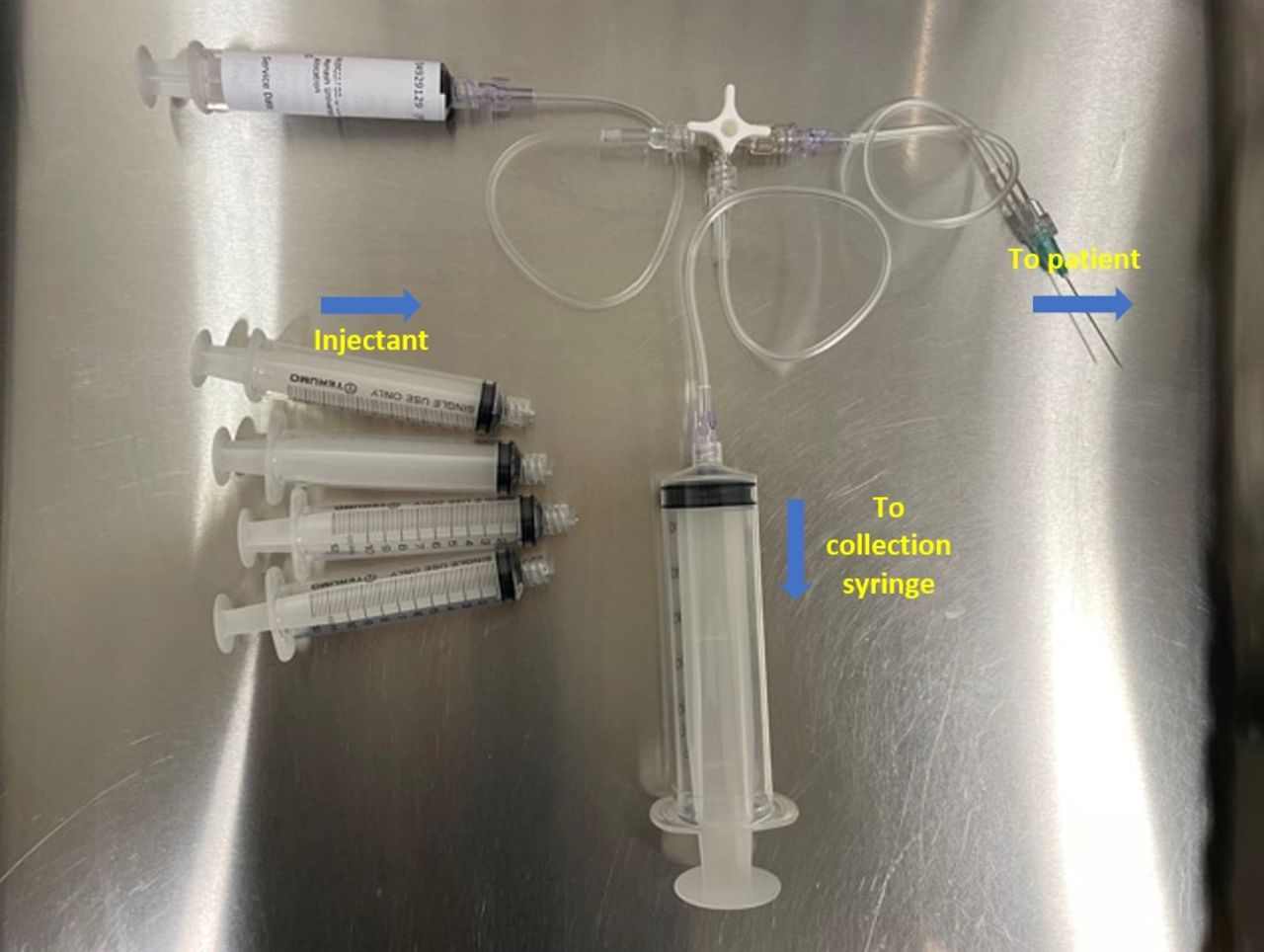
Three-way stop cock is used to divert the injectant into the 50 mL collection syringe for the sham group.
Unblinding will only be permissible if one of the participants experiences a serious adverse event that may be related to one of the medications administered. If this occurs, a member of the trial management team (who is not involved in data collection) will unblind the participant and notify the treating medical staff.
Three injection groups
In participants with bilateral symptoms, the most symptomatic side (volunteered by the patient on questioning, or the right side if they cannot define the more symptomatic side) will be injected. All participants will receive a single injection (see figure 3) that will be delivered by an experienced musculoskeletal radiologist while participants are lying prone. Immediately post-injection, participants will be asked which injection they believe they received (they are able to answer whether they think it is intervention, sham or that they do not know32) to assess the success of participant blinding.
{kind=link}
{kind=link}
{kind=link}
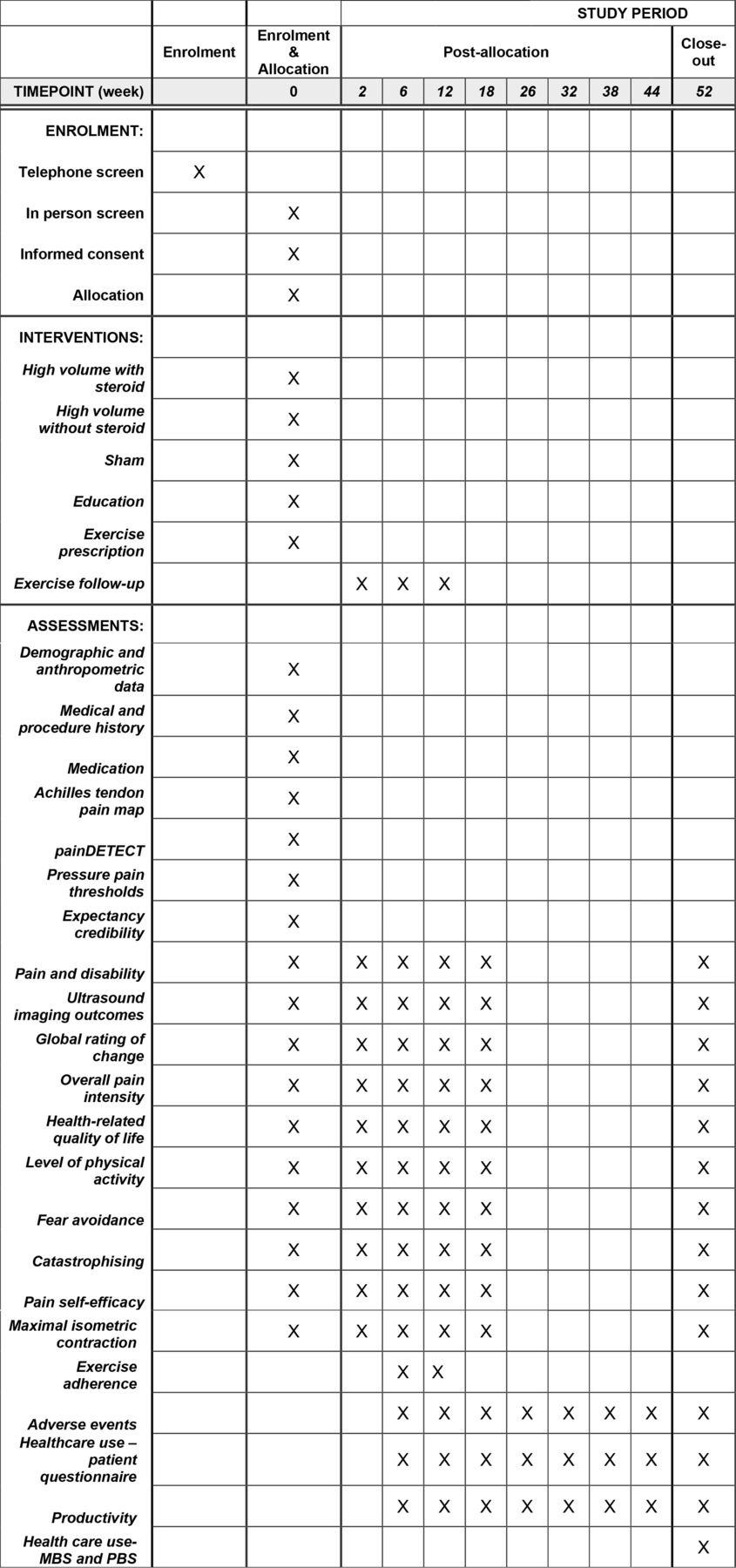
Schedule of enrolment, interventions and assessments.
High-volume injection with corticosteroid: The HVI will be injected into the interface between Kager’s fat and the Achilles tendon. The local anaesthetic and the corticosteroid is injected followed by the four saline syringes by using a connecting tube (allows consecutive syringes to be attached). The position of the needle will be monitored continuously by ultrasound and the needle will be moved gently across the anterior aspect of the tendon to ensure uniform effect over the pathological area.
Tendon sheath injection without corticosteroid: This injection is identical to the HVI with corticosteroid, but the first syringe will only contain exactly 10 mL of 0.5% bupivacaine hydrochloride (no corticosteroid).
Sham injection: The sham injection consists 2–3 mL of 0.5% bupivacaine hydrochloride and will be injected under image guidance by the same expert radiologist, deep to the tendon and away from the interface between Kager’s fat and the Achilles tendon (at last 10 mm away). The sham injection will result in a numbing effect, like the HVI.
Post-injection treatment
Immediately after the injection, an appropriately trained researcher (figure 1) who is blind to treatment allocation will deliver identical education and a 12-week exercise programme to all participants (see table 2 Figure 3), based on prior HVI work.20 25 This includes an evidence-based and progressively loaded exercise protocol for AT,33 34 education about physical activity modification and resumption, and advice about progressing and regressing exercise load, based on a pain monitoring model.35 The researcher will also provide education about AT (pathology, pain, risk factors, prognosis, treatments, recovery times) and post-injection advice. The information provided to patients (in printed form) at baseline is shown in online supplemental file 1 and the exercise videos provided are shown in online supplemental file 2. Participants will be reviewed by the researcher on four follow-up occasions—1 week (brief phone call to check post-injection response and whether exercise was commenced), 2 weeks via teleconference and 6 weeks in person (modify the exercise as required, eg, progress load, modify technique), and at 12 weeks—to provide evidence-based advice about continuing exercise beyond the intervention period.25
To ensure participants are supported and to optimise uptake of the 12-week exercise programme, they will be able to email or call the researchers with questions and will be provided with custom-produced basic online exercise videos and education. Participants will be advised to refrain from using other physical therapy interventions and non-steroidal anti-inflammatory medications but will be allowed to take paracetamol (up to 4 g/day) for pain relief as required.
Intervention fidelity
The fidelity of each intervention (injections, education and exercise) will be monitored at regular intervals (at least every 8 to 10 injections (minimum of 19 assessments throughout the trial)) by a member of the trial management group who is not involved in data collection or analysis. Radiologists and physiotherapists delivering the treatment will be provided with feedback and strategies to improve fidelity as appropriate.
Outcome measures
Outcomes are either self-report or performed by appropriately trained researchers or ultrasonographer blinded to the treatment allocation. Outcome assessment will occur at baseline, 6, 12, 26 and 52 weeks (see figure 3). All follow-ups will occur at the same imaging facility as baseline assessment. All data collected will be pseudoanonymised (retaining code file) and stored on a secure cloud-based server. Participants will complete self-report outcomes in relation to their most symptomatic Achilles tendon if they have bilateral AT.
Primary outcome: Pain and function will be assessed with the VISA-A, a well-validated, reliable and disease-specific tool.36 This outcome includes pain, function and activity domains that are clinically relevant to patients with AT. Scores range from 0 to 100, with 100 indicating no symptoms or function/activity limitations. The final two question of the VISA-A were designed for sporting populations and will be modified in our study to relate to our mixed sporting and non-sporting population. Question 7 will be modified from “Are you currently undertaking sport or other physical activity?” to “Are you currently undertaking sport or other physical activity, including walking?”. Question 8 asks about pain and disability during Achilles tendon loading sport. ‘Achilles tendon loading sport’ will be changed to ‘weight-bearing activity’. Both versions of question 8 will be included so that responsiveness of both versions of the VISA-A can be evaluated.
Secondary outcomes:
Achilles tendon thickness and Doppler signal: assessed with ultrasound imaging using reliable methods.37 Doppler signal will be assessed by quantifying pixel count.38 This outcome is important to assess whether tendon adaptation and change in Doppler signal is a potential mechanism of the HVI.
The Global Rating of Change Scale: an 11-point scale in which the participant is asked to rate their perceived overall change in their AT condition from the time that they began the study until the present, as Worse, No Change or Better. If they indicate worse, the patient will then be asked how much worse on a 5-point scale from Very Much Worse to Slightly Worse, and if they are better, they will be asked how much better on a 5-point scale from Slightly Better to Very Much Better.39
Overall pain intensity: measured using the 100 mm visual analogue scale (VAS), participants will rate the worst pain during the last week (zero=no pain; 100=worst pain possible).
Health-related quality of life: measured with the EQ-5D-5L, a validated and reliable tool,40 including five domains (mobility, self-care, usual activities, pain/discomfort and anxiety/depression), and a rating of overall health state from 0 (worst health state imaginable) to 100 (best imaginable health state) using a VAS.
The level of physical activity in the previous week: evaluated using the 7-day Recall Physical Activity Questionnaire, a valid and reliable tool.41 Participants will be asked to recall time spent sleeping and doing physical activity (work, leisure, household activities) over the past 7 days.
Fear avoidance and catastrophising: the Tampa Kinesiophobia Scale42 and Pain Catastrophising Scales43 are validated questionnaires used to measure fear avoidance and pain catastrophising, respectively.
Pain self-efficacy: assessed with the pain self-efficacy questionnaire (PSEQ).44 The PSEQ measures how confident a patient is in undertaking a range of activities despite their pain.
Maximal voluntary isometric contraction (MVIC) and rate of force development (RFD)45: seated calf raise RFD and MVIC. Standard strength (MVIC and RFD) tests will be performed on a custom chair equipped with a force plate that measures plantarflexor force. Participants will have an adequate warm-up (10 repetitions of calf raises with the knee bent and knee straight) and then perform 4 practice and 2 recorded trials. The instructions will be “push as hard and fast as possible”.
Other secondary outcomes: selected outcomes will be assessed at every 6 weeks from weeks 6 to 52 via online questionnaire (if not coinciding with an assessment visit time).
Adverse event: an adverse event is defined as any unfavourable or unintended diagnosis, sign, symptom, or disease associated with the study which may or may not be related to the intervention (eg, tendon rupture, fall, injury or change in medical status) and prevents participation in the intervention for at least seven consecutive days. Participants will record what each adverse event was (eg, tendon rupture), impact on their ability to undertake the intervention and for how long.
Serious adverse events: defined as an event related or not related to the intervention that results in death, life-threatening complication, hospitalisation, surgery, permanent or temporary physical disability, congenital abnormalities, or any findings the CI and/or research team feel can lead to significant health hazards. Examples of serious adverse events include but are not limited to surgery for tendon rupture and heart attack requiring overnight hospitalisation.
Reporting of adverse events
If the adverse event is not defined as serious, the adverse event is recorded in the trial file and the participant is followed up by the research team. All serious adverse events will be recorded in the participant’s trial file and reported to the chief investigator immediately. The chief investigator will let the trial sponsor know within 24 hours, as well as data monitoring, trial management and steering committees (described later).
Action plan for addressing adverse events
If the adverse event occurs at the trial setting (IOP, Melbourne), then the centre policy for adverse events will be followed. If the adverse event occurs outside the trial setting, then the participants will be advised to see their GP or go to the emergency department for appropriate management. All the treatment costs will be covered by the sponsor’s insurance.
b. Exercise adherence: participants will record the number of exercise sessions completed each week over the previous 4 weeks (adherence is the percentage of prescribed sessions that are completed). Adherence will be categorised as (i) poor, <25%; (ii) moderate, 25% to 50%; (iii) good, >50% to 75%; or (iv) excellent, >75%.46 Exercise adherence will be measured at 6 and 12 weeks.
c. Healthcare use: Medicare Benefits Schedule (www.mbsonline.gov.au/) and Pharmaceutical Benefits Schedule (www.pbs.gov.au) data will be extracted to measure use of subsidised healthcare services. The use of other health service and co-interventions will be measured by asking for a yes/no response followed by a phone call.
Productivity: productivity (including absenteeism and presentism) will be measured using the ‘iMTA Productivity Cost Questionnaire’.47
Statistical methods
Data will be double entered to minimise errors and coded to ensure blinding of the statistician undertaking the analyses. In participants with bilateral symptoms, the most symptomatic side (or the right side if they cannot define the more symptomatic side) will be analysed to maintain independence of data. Statistical tests will be two-tailed with statistical significance level set at 0.0167 for each of the three pairs of comparisons of trial arms. We will also analyse the primary outcome with alpha set at 0.05 and provide this as a supplemental file for future systematic reviewers. All randomised patients will be included in the analysis (ie, intention-to-treat) for primary and secondary outcomes (except for safety outcomes). We will also undertake secondary per-protocol analyses. Demographic characteristics (eg, age, gender) and other baseline measurements (eg, duration of symptoms) will be reported by treatment arm. Between-group differences in the primary and secondary outcomes measures will be compared at 12, 26 and 52 weeks, with the primary outcome being at 52 weeks. Continuously scored outcome measures will be analysed using linear mixed models with adjustments for baseline scores and variables that are found to influence the outcome. Ordinal scaled data will be analysed using non-parametric tests and modelled with proportional odds regression adjusted for repeated assessment of subjects. Dichotomous outcome measures will be compared using relative risk, risk difference and number needed to treat using generalised estimating equations. Sensitivity to missing data will be assessed using multiple imputation models incorporating predictive baseline and post-baseline variables.
Economic analysis
Alongside the main trial, there will be a parallel, trial-based cost-utility analysis. This economic analysis will consider the incremental cost of each of the intervention conditions compared with the sham for gaining one quality-adjusted life year. Quality-adjusted life years will be calculated using an area under the curve approach on EQ-5D-5L utility scores collected at each assessment. These scores will be generated using the Dolan cross-walk approach.48 A societal perspective will be taken by calculating healthcare and productivity costs incurred by participants. Valuation of health services costs will be based on standard rates published by the Australian government (National Weighted Activity Unit costs for hospitalisations (The Independent Hospital Pricing Authority website, https://www.ihpa.gov.au/what-we-do/national-weighted-activity-unit-nwau-calculators-2015-16), Medical Benefits Schedule and Pharmaceutical Benefits Schedule prices for other subsidised health costs, market prices for other costs). Bootstrap resampling will be used to generate CIs for cost-utility estimates and for construction of acceptability curves. One-way sensitivity analyses will also be conducted to examine the impact of variation in key analysis inputs.
Trial governance
A trial management group (TMG) will be convened and meet up to quarterly. In addition to this, a data safety management board (DSMB) and a trial steering committee (TSC) will also be convened. The TMG is composed of the research team and will meet quarterly to oversee trial management. The DSMB will include an independent multidisciplinary group consisting of two biostatisticians and clinical experts (injection and exercise), who will monitor the safety and compliance issues related to the trial. The TSC will include a core member of the TMG, expert clinicians (injections and exercise), and at least one lay representative and will be responsible for issues such as recruitment, trial management and safety concerns. The TSC and DSMB will meet once to twice a year. The first review of safety (undertaken by the full TMG) will occur when 20 participants have completed the 12-week assessment and then every 6 months until the conclusion of the trial.
Patient and public involvement
Patients will be involved in the trial TSC from inception and will have a role in developing the research design, outcome measures and recruitment strategy.
Ethical considerations
This study protocol was granted ethics approval by the Monash University Human Ethics Committee (no: 13138). Any amendments to the protocol will be reviewed by this ethics committee and communicated to the trial funder and trial registry.
Dissemination
The findings of this trial will be presented at national and international conferences and submitted to a peer review journal for publication. Dissemination assets will be created, including audience-specific media releases and infographics, and disseminated to stakeholders including patients and clinicians.
Discussion
AT is common and disabling. As many as up to 44% of people with AT fail to recover with conservative first-line treatment13 37 49 and are offered other treatments including injections.14 Despite the common use of injections, there are currently no injection treatments for AT with demonstrated efficacy. Although corticosteroid injections have demonstrated short-term efficacy, there may be recurrence of symptoms for some people in the longer term.15 The lack of an efficacious injection for AT may prolong disability for many people who are affected and significantly increase healthcare use and costs. HVI may be an effective treatment for AT but has not been adequately tested. The proposed trial will address this knowledge gap by evaluating the efficacy of the HVI with and without corticosteroid versus sham for AT over 12 months. This research has the potential to enhance recovery of patients thereby improving clinical outcomes and reduce societal costs by reducing failed injection treatments and ongoing disability.
Limitations
The radiologist is not blind to the sham interventions, but we will implement measures including a standardised script to ensure that all participants have comparable interventions with this caregiver. It is possible that people who have had a corticosteroid injection close to 3 months ago and up to a year prior may still have long-term adverse events from this injection that influences our findings. These effects are likely to be equal across groups given the randomisation process. A potential limitation to external validity is the single-centre design; however, this is appropriate for the efficacy paradigm adopted in this trial.
Data availability statement
Data will be made available on request at the completion of the trial.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors All authors were involved in developing the methods for this trial and writing the protocol manuscript. PM is the guarantor.
Funding This research has been funded by a National Health and Medical Research Council (NHMRC) Project Grant (APP1164268).
Disclaimer The funder does not/will not have any role in study design, data collection, analysis or interpretation for this trial.
Competing interests DC is an interventional radiologist who receives pay for interventional procedures such as Achilles tendon injections as part of his work. MU is chief investigator or co-investigator on multiple previous and current research grants from the UK National Institute for Health Research, Arthritis Research UK and is a co-investigator on grants funded by the Australian NHMRC. He is an NIHR Senior Investigator emeritus. He has received travel expenses for speaking at conferences from the professional organisations hosting the conferences. He is a director and shareholder of Clinvivo Ltd that provides electronic data collection for health services research. He is part of an academic partnership with Serco Ltd, funded by the European Social Fund, related to return to work initiatives. He is a co-investigator on three NIHR-funded studies receiving additional support from Stryker Ltd. He has accepted honoraria for teaching/lecturing from consortium for advanced research training in Africa. Until March 2020, he was an editor of the NIHR journal series, and a member of the NIHR Journal Editors Group, for which he received a fee. RSK is chief investigator or co-investigator on multiple previous and current research grants from the UK National Institute for Health Research (NIHR) and Arthritis Research UK member. RSK is a member of the UK National Institute for Health Research (NIHR) Health Technology Assessment Clinical Evaluation and Trials board, NIHR Integrated Clinical Academic Doctoral panel, chair of the NIHR Research for Public Benefit board and holder of a NIHR Fellowship award.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.